Research Group Molecular Drug Design (Head: Univ.-Prof. Dr. G. Wolber)
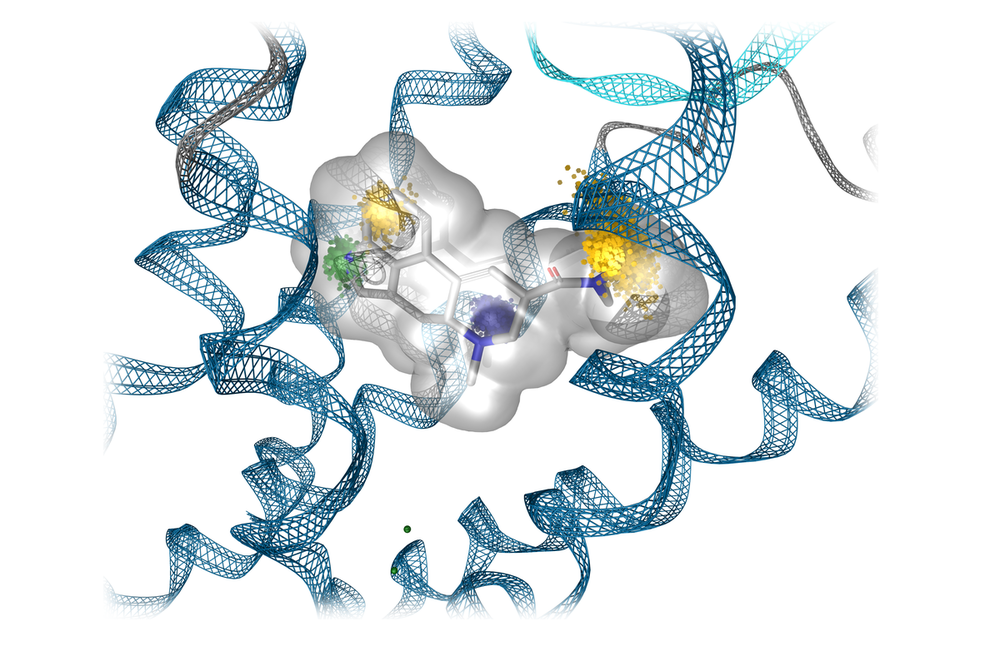
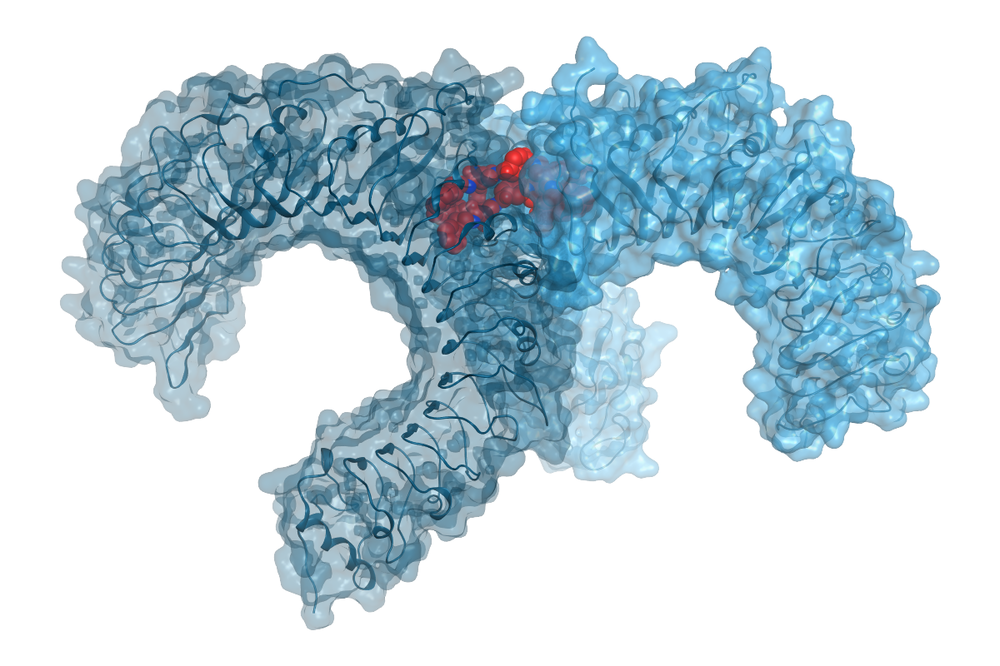
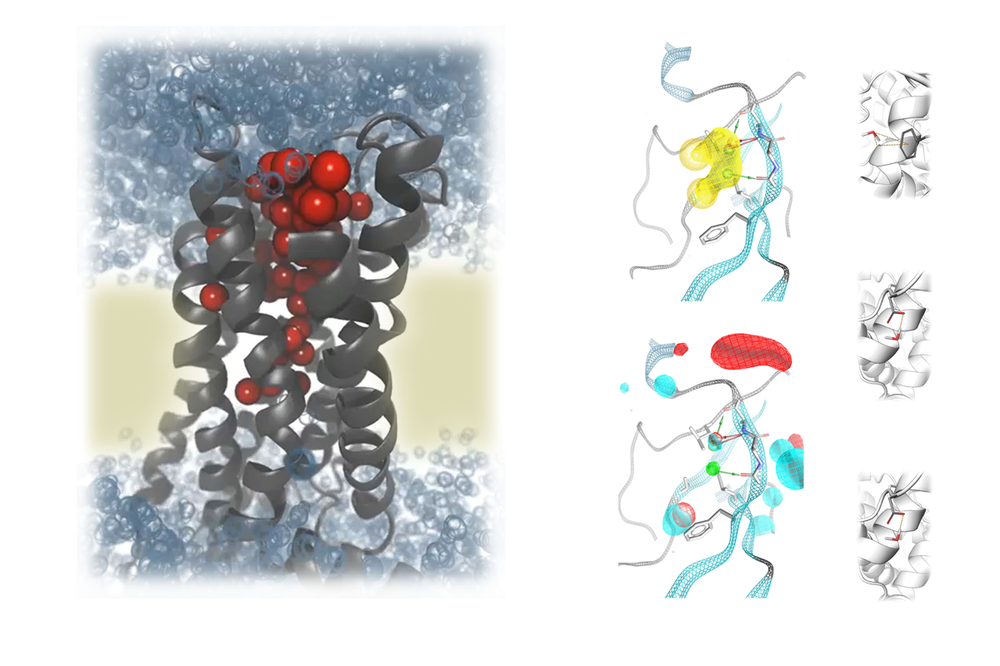
The research focus of the working group Molecular Drug Design headed by Univ.-Prof. Gerhard Wolber is the discovery, design, development, and synthesis of new bioactive molecules as well as the biophysical and biochemical characterization of their binding to target molecules and synthesis. The computer-aided molecular design of active compounds plays a central role in our methodology, both structure-based and ligand-based, through modern machine learning methods.
Our research projects focus on (new) data-oriented methods and the application of artificial intelligence to improve virtual screening approaches, rational synthesis strategies, and the mechanistic investigation of protein-ligand interactions. Interdisciplinarity is an integral part of our research. The development of bioactive molecules requires cooperation with other subjects in pharmacy, biology, analytics, structure elucidation, and medicine. This interdisciplinarity represents one of the great strengths of pharmacy and lays the foundation for translational research.
We focus on rational drug design in the following areas:
- Development and application of new computer-aided methods in drug design
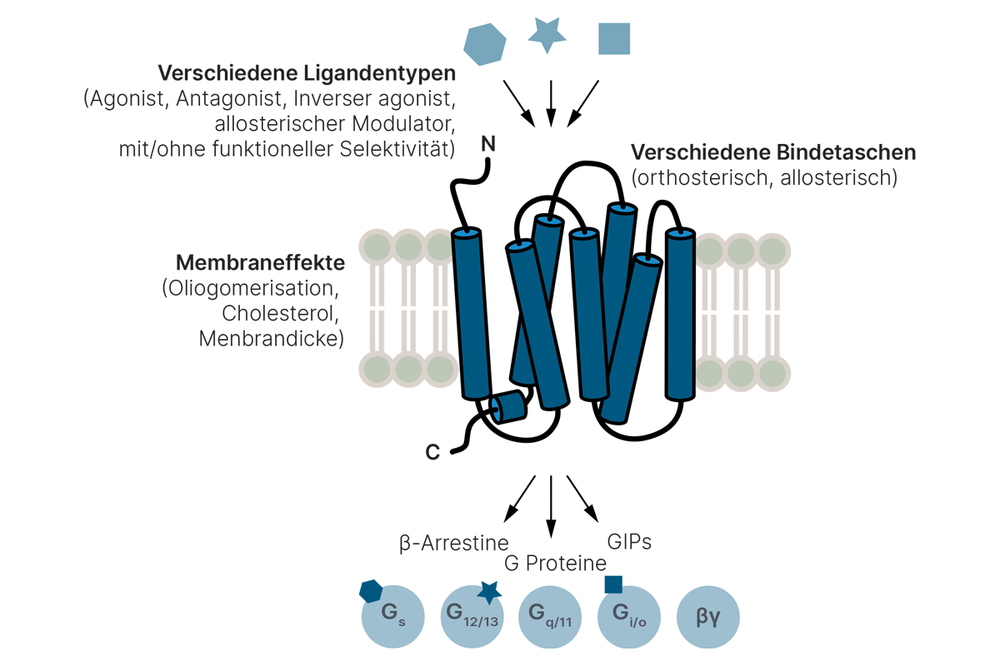
- Tailored ligands with functional selectivity for G protein-coupled receptors
- Toll-like receptors: modulation of the innate immune response for inflammation regulation and cancer therapy
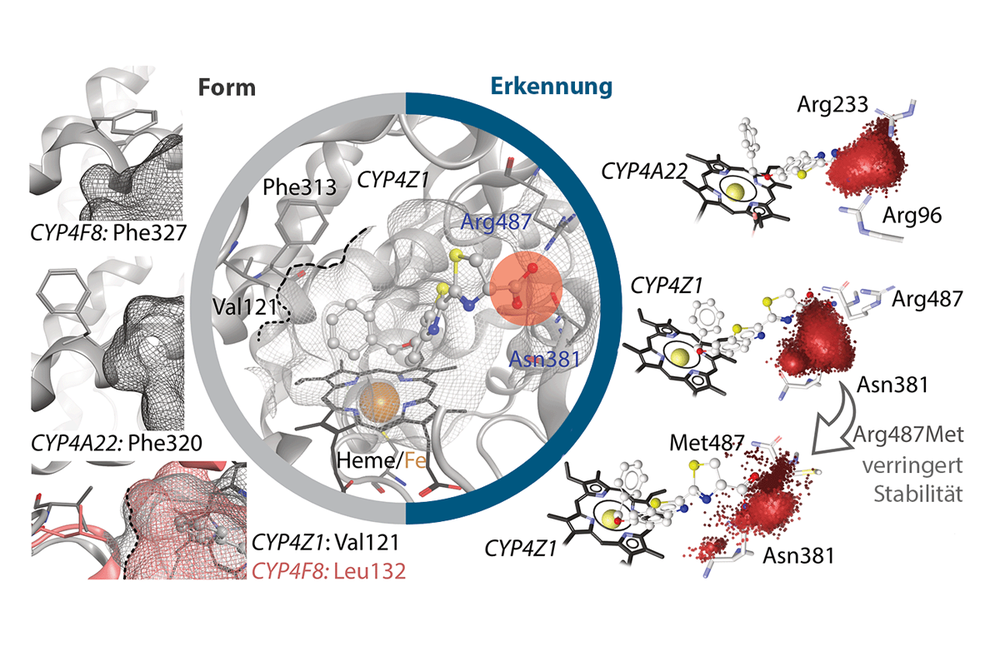
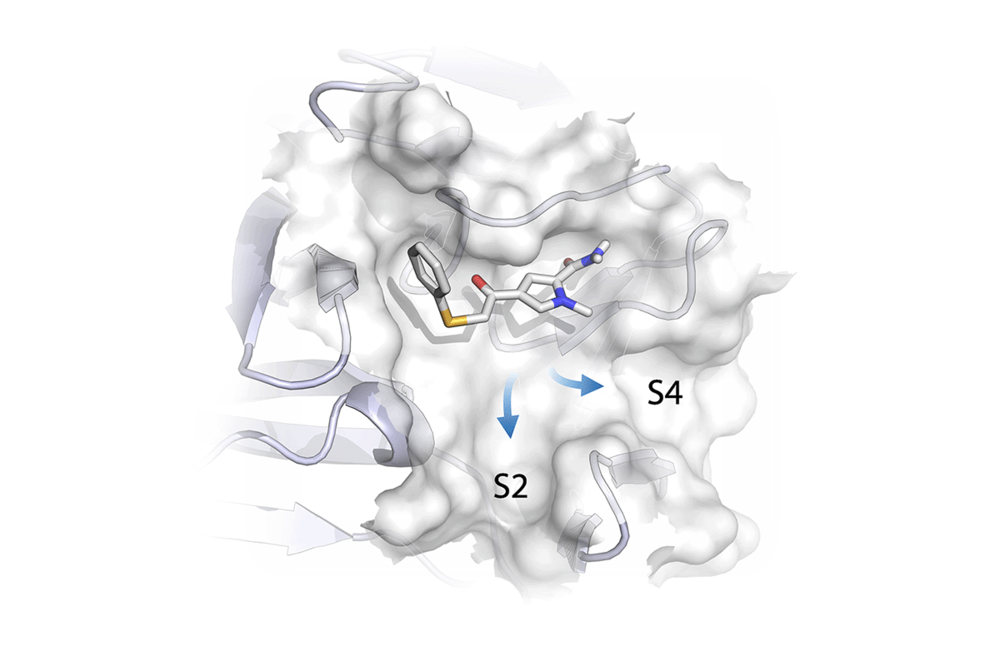
- Cytochrome P450 enzymes: Metabolism & Applications in Cancer Therapy
- Viral protease inhibitors by fragment-based de novo design