
New Publication in PCCP
Structure investigations of peptides using DFT, IRMPD and IM-MS
News from Feb 24, 2015
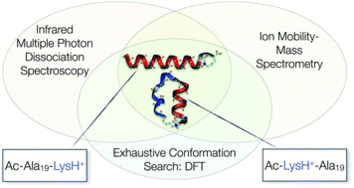
Exploring the conformational preferences of 20-residue peptides in isolation: Ac-Ala19-Lys + H+ vs. Ac-Lys-Ala19 + H+ and the current reach of DFT
Reliable, quantitative predictions of the structure of peptides based on their amino-acid sequence information are an ongoing challenge. We here explore the energy landscapes of two unsolvated 20-residue peptides that result from a shift of the position of one amino acid in otherwise the same sequence. Our main goal is to assess the performance of current state-of-the-art density-functional theory for predicting the structure of such large and complex systems, where weak interactions such as dispersion or hydrogen bonds play a crucial role. For validation of the theoretical results, we employ experimental gas-phase ion mobility-mass spectrometry and IR spectroscopy. While unsolvated Ac-Ala19-Lys + H+ will be shown to be a clear helix seeker, the structure space of Ac-Lys-Ala19 + H+ is more complicated. Our first-principles structure-screening strategy using the dispersion-corrected PBE functional (PBE + vdWTS) identifies six distinctly different structure types competing in the low-energy regime (≈16 kJ mol−1). For these structure types, we analyze the influence of the PBE and the hybrid PBE0 functional coupled with either a pairwise dispersion correction (PBE + vdWTS, PBE0 + vdWTS) or a many-body dispersion correction (PBE + MBD*, PBE0 + MBD*). We also take harmonic vibrational and rotational free energy into account. Including this, the PBE0 + MBD* functional predicts only one unique conformer to be present at 300 K. We show that this scenario is consistent with both experiments.
go to article DOI: 10.1039/C4CP05541A



