
Research
Alternative splicing multiplies the number of possible proteins generated from a single pre-mRNA and therefore is one of the most abundant mechanisms to control protein expression and function posttranscriptionally. Indeed, above 90% of human multi-exon pre-mRNAs are alternatively spliced, many of them in a tissue-, differentiation- or activation-status dependent manner. Alternative splicing does not only lead to differential isoform expression, but can, through the inclusion of premature termination codons, also induce nonsense-mediated decay (NMD) to control the overall abundance of mRNAs.
Our current work is focused on a mechanistic understanding how body temperature changes, for example in a circadian manner, during ageing, or during therapeutic manipulation, control alternative splicing and gene expression and how this impacts on functionality.
In the past years, we have pioneered research that connects changes in body temperature with kinase activity, global control of alternative splicing and gene expression through NMD and functional relevance. We have for example shown that subtle changes in body temperature control the antiviral immune response, which may contribute to explain why older individuals with reduced body temperature are less well protected against some viral infections. In addition, we have used our mechanistic understanding of body temperature-controlled alternative splicing coupled to NMD to developed ASOs that increase expression of the broadly neuroprotective protein RBM3 at normothermia (‘hypothermia in a syringe’). We are currently developing these ASOs as therapeutic approach in hypoxic ischemic encephalopathy (HIE) and other neurodegenerative conditions.
Some of our future goals are to identify additional therapeutically relevant targets of hypo- and hyperthermia, to understand the mechanistic basis of their temperature-regulation and to use this mechanistic understanding to manipulate their expression at normothermia as new therapeutic concept.
In an independent line of research, we use the secretory pathway as a model system to study the impact of alternative splicing on a fundamental cell biological process. We have discovered the first connection of alternative splicing and the secretory pathway and showed that activated T cells increase their secretory capacity through a splicing-based adaptation of the COPII machinery (Wilhelmi et al., Nat Comm, 2016). We then went on and combined bioinformatics with cell biology to provide the first global view of how alternative splicing controls membrane trafficking (Neumann et al, JCS, 2019). More recently, we have started to address the role of alternative splicing of members of the COPII coat on the secretion of large cargo such as collagen and chylomicrons. (Ostwaldt et al., RNA, 2025)
The body temperature of homeothermic organisms is tightly controlled in a narrow range. However, we have shown that subtle fluctuations in the core body temperature e.g. due to circadian temperature oscillations, hypothermia or fever, result in global changes in alternative splicing and gene expression.
To address the functionality of body temperature controlled alternative splicing and gene expression we have used extensive analyses of RNA-Seq datasets. Interestingly, we find cancer-associated genes to be differentially expressed and spliced at different temperatures, which may result in a tumor-suppressive environment at higher temperature. We are now addressing the mechanistic basis and functional consequences of temperature-controlled expression of oncogenes and tumor suppressors, which may contribute to a better molecular understanding of thermotherapy as a therapeutic approach.
RNA-Seq and downstream bioinformatics allow the transcriptome-wide quantification of splice site choices. By integrating multiple RNA-Seq datasets from siRNA-mediated perturbations of core components of the spliceosome, we have defined groups of spliceosomal proteins implicated in specific changes in alternative splice site choice. A multi-level bioinformatics analysis pipeline revealed common patterns or features within changed splicing events and led to our current focus on NAGNAG alternative splicing. We now use classical biochemical experiments to gain mechanistic insights into protein-specific changes in splice site selection. After careful validation of bioinformatics predictions using splicing-sensitive RT-PCRs we use bioinformatics-based mutations of cis-regulatory RNA sequences or structure-guided mutations of trans-acting factors for a detailed mechanistic analysis of alternative splice site selection. Ultimately, the combined bioinformatics and biochemical approaches will reveal new paradigms of splice site selection and contribute to understand the incredibly complex regulation of (alternative) splicing.
![]()
A, Heatmap confirming specific splicing factor knockdowns within RNA sequencing data.
B, Volcano blot revealing a directed effect of a splicing factor knockdown on alternative acceptor splice site choice (significant events are highlighted by colors).
C, Correlation matrix reveals co-regulation of a certain intron subtype by a subset of splicing factors.
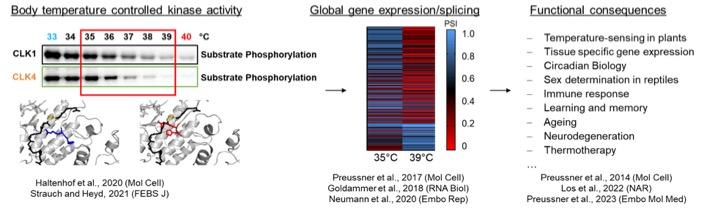
Our work on temperature-controlled alternative splicing and gene expression started when we found that alternative splicing of U2AF26 is controlled in a time-of-the-day dependent manner and that this splicing event plays a role in resetting the circadian clock in vivo. This is the first functionally well-characterized rhythmic alternative splicing event in a mammalian system (Preussner et al., Mol Cell, 2014). We then set out to determine cis-acting elements and trans-acting factors that control rhythmic alternative splicing. We found that circadian changes in body temperature act as systemic signal to globally control alternative splicing and gene expression through regulating the phosphorylation status of SR proteins (Preussner et al., Mol Cell, 2017). Next we showed that the activity of a family of kinases, CLKs, is extremely temperature-sensitive in a narrow temperature range due to conformational changes in the active center and that this leads to reduced kinase activity at the upper end of the physiologically relevant temperature in diverse organisms, suggesting, amongst many other functional consequences, a connection to temperature-dependent sex determination in reptiles (Haltenhof et al., Mol Cell, 2020). Altered CLK activity then translates into global changes in alternative splicing through regulating the phosphorylation status of SR proteins. Interestingly, we have also shown that many temperature-controlled alternative splicing events control poison exons that induce nonsense-mediated decay (AS-NMD), thus providing a means how temperature-controlled alternative splicing can also control gene expression. In fact, AS-NMD plays a substantial role in controlling the temperature-dependent transcriptome (Neumann et al., Embo Rep, 2020), which has diverse functional consequences, some of which we are currently analyzing. For example, we have recently shown that subtle changes in body temperature alter STAT2 levels through AS-NMD, thereby controlling the antiviral defense. This finding may contribute to explain why older individuals with lower body temperature are more susceptible to severe Sars-CoV-2 infection than children (Los et al., NAR, 2022). With this work, we have characterized the full cascade, from a body temperature sensor to altered phosphorylation of SR proteins, a global change in alternative splicing and gene expression through AS-NMD and a clinically relevant functional consequence. Finally, we have discovered the long-elusive mechanism that controls cold-induced expression of the neuro- and cytoprotective protein RBM3. This solely depends on AS-NMD, which leads to degradation of the RBM3 mRNA at warmer temperature (already at 37°C). We have developed ASOs that target the poison exon of RBM3 to inhibit NMD and induce its expression at normothermia. These ASOs are highly neuroprotective in a mouse model for prion disease and hold immense potential to be developed for use in humans in diverse conditions from neonatal hypoxic ischemic encephalopathy to Alzheimer’s disease (Preussner et al., Embo Mol Med, 2023).