
STED microscopy, a recent advance in the analysis of supra-molecular architectures
Cellular membranes display a high degree of order to spatio-temporally organize signaling. Local control over the assembly of macromolecular complexes is likely to be a key process here. However, classical approaches as biochemistry are not always suited to resolve the composition and dynamics of these "cyto-architectures" in spatio-temporal terms.
To take the example of synapses (see below), most structures relevant for a deeper understanding of active zones (e.g. synaptic vesicles and dense bodies) are at or below 100 nm in size. Owing to their diffraction limited resolution, confocal and epifluorescence microscopes often cannot resolve these "cyto-architectures". Electron microscopy, on the other hand, provides sufficient resolution, but the required labeling efficiency has hitherto not been achievable.
Recent advances in optical physics have shown that the diffraction barrier of far-field fluorescence microscopy can be broken by stimulated emission depletion (STED) (Hell, 2003; Klar et al., 2000; Willig et al., 2006a; Willig et al., 2006b). In a typical STED microscope the excitation beam is overlapped with a doughnut-shaped beam that is capable of de-exciting fluorophores by stimulated emission. Co-alignment of the beams ensures that fluorescence is allowed only in the central area of the excitation spot where the doughnut beam is close to zero (Fig. 1, left panel). Scanning with a narrowed spot across the sample yields subdiffraction images. With a sufficiently intense doughnut, the fluorescent spot of a STED microscope can be sharpened down to the molecular scale (see figure 1 illustrating the principle of STED taken from Hell et al., 2004).
Recently, the company Leica has developed a STED microscope ("Leica TCS STED") based on a robust architecture for STED laser alignment. We have been using and adapting this instrument for biological application with Leica. The Leica STED microscope will have a specified optical resolution of 90 nm fluorescence width at half maximun (FWHM). In our hands, 70 nm were typically reached at the prototype throughout our experiments with Leica in Mannheim. Even smaller objects can be resolved due to the very steep otpical transfer function of the STED microscope, e.g. allowing the clear separation of 65 nm beads (not shown).
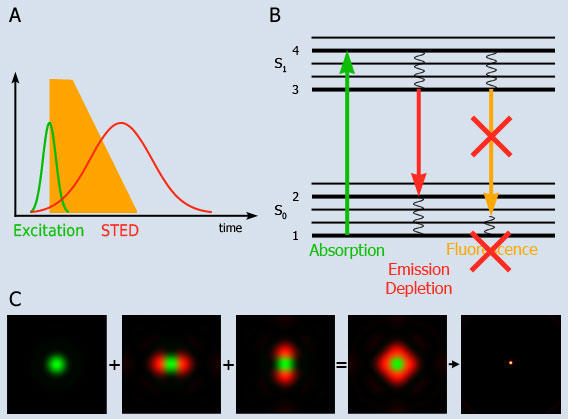
Fig. 1: Principle of STED-microscopy
A: A short excitation pulse (<1 ps) leads to fluorescence which is directly quenched by a long STED pulse (10-200 ps).
B: Molecules are excited into the fluorescent state S1 from where they normally return to the ground state S0 by spontaneous fluorescence emission, yet depletion of the fluorescence can be optically enforced. Stimulated emission depletion is achieved by intense light pulses with durations shorter than the S1 lifetime. As a result no fluorescence takes place in the depleted region.
C: The excitation laser spot is shown in green, which is surrounded by the STED laser shown in red. The doughnut shaped sum of the STED overlaps with the the diffraction limited excitation spot. The STED quenches fluorescence except in the innermost area, which leads to an excitation spot of subdiffraction dimensions.
Analysis of Bruchpilot using STED: an entry point into active zone molecular architecture
Active zones are the sites where the fusion of transmitter-filled vesicles takes place. While further insights into the molecular composition of these structures would be of importance for plasticity, learning and memory as well as nervous system disease processes, still we lack an integrated understanding of the molecular machinery constituting active zone structure as well as its function for the control of synaptic vesicle release. As already mentioned, the fact that active zones are typically only about 200 nm in diameter has so far restricted the light microscopic analysis of their substructures. We have now started to apply the power of STED microscopy to further resolve active zone organization. As pointed out above, we recently identified the Bruchpilot protein (BRP), highly related to the mammalian CAST/ELKS family of active zone proteins, as a central organizer of active zone structure and function in Drosophila (Kittel et al., 2006; Wagh et al., 2006; see figure 2). Active zones are characterized by voltage-gated Ca2+ channels, docked vesicles, and electron-dense structures, which are referred to as T-bars at Drosophila synapses. In the absence of BRP, T-bars were entirely lost and evoked vesicle release was severely impaired, due to a defect in the clustering of presynaptic Ca2+ channels.
STED fluorescence microscopy has revealed that the monoclonal antibody Nc82, which recognizes the C-terminus of BRP, labels a doughnut-shaped distribution at active zones (Fig. 2; Kittel et al., 2006). Further analysis using N- and C-terminal antibodies for the BRP protein showed an extended conformation and a "funnel-like" organization of the protein; the C-terminal and N-terminal signal hardly overlap spatially. Instead, the N-terminal label is found about 100 nm closer to the plasma membrane than the C-terminal label.
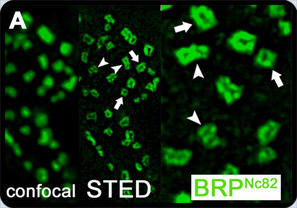
Fig. 2: Unlike confocal, STED microscopy revealed doughnut-shaped structures recognized by monoclonal antibody Nc82. Viewed from above, both single (white arrows) and clusters of multiple rings (arrowheads) were identified (Taken from Kittel et al., 2006).
Further references not listed within our publications:
- Hell SW (2003): Toward fluorescence nanoscopy. Nat Biotechnol 21(11):1347-55.
- Hell SW, Dyba M, Jakobs S (2004): Concepts for nanoscale resolution in fluorescence microscopy. Curr Opin Neurobiol 14(5):599-609.
- Klar TA, Jakobs S, Dyba M, Egner A, Hell, SW (2000): Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. PNAS 97(15):8206-10.
- Willig KI, Kellner RR, Medda R, Hein B, Jakobs S, Hell SW (2006a): Nanoscale resolution in GFP-based microscopy. Nat Methods 3(9):721-3.
- Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW (2006b): STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature 440(7086):935-9.
More infomation is available about the background of our research as well as about ongoing projects. Click here for a summary of our research.
