Research
Introduction
The group develops new methods for the identification and optimization of protein ligands by fragment ligation and fragment ligation assays. Aim of these works is a more profound understanding of the contributions of molecular substructures to protein-ligand recognition. Cooperative enhancement of fragment-interactions is in the focus of our interest.
Progress in ligand identification is translated into optimized chemical probes which are used for the structural and functional characterization of proteins and for the visualization of molecular interactions in living systems. In special cases the chemical probes are starting points for investigating the pharmacological potential of the developed bioactive molecules.
Most protein targets of the group are disease-related enzymes including proteases and phosphatases relevant to clinical indications including cancer, Alzheimer, tuberculosis, and SARS. Recently, the targeted proteins have been extended towards receptors and protein-protein interactions.
Fragment-based ligand discovery
„Molecular fragments“ are defined as low-molecular weight building blocks (M ≤ 250 Da). Thus they are in the size range of a protein binding pocket hosting a single amino acid side chain. Fragment-based drug discovery consists of the step-wise construction of high-affinity protein ligands from low-affinity fragments employing fragment combinations. Linking of fragments creates hetero-multivalent ligands in which binding of the monovalent fragments can be enhanced cooperatively. Thus, fragment-based ligands often display higher ligand efficiencies and comply better with pharmacokinetic parameters than ligands obtained from high-throughput screening and/or natural product libraries.
Dynamic Ligation Screening
Classically, low-affinity fragments have been identified by biophysical methods such as NMR and X-ray cristallography. We have developed a novel principle for the detection of fragment binders, denominated as Dynamic Ligation Screening. DLS is an approach capable of measuring the activity of reversibly formed fragment combination in a biochemical assays. Thus, DLS enables the spatially resolved detection of low-affinity fragments in high-throughput bioassays. Subsequently, the detected fragments can be linked irreversibly to yield high-affinity protein ligands.
For example, in the first iterative application of the method, a low-affinity peptidic inhibitor of SARS protease has been transformed into a highly active, non-peptidic molecule.
Based upon the DLS-methodology ligands for several protein targets have been developed. Starting from various proteases, meanwhile protein tyrosin phosphatases and protein protein interactions have been used as targets for fragment-based ligand development.
The initial assay concept has been diversified considerably: First, ligated fragments were detected by competition with a fluorogenic substrate („substrate-competition“). Next the approach was extended towards binding assays. In this, the competition of ligation products with a fluorescently labelled binding partner was investigated by using fluorescence anisotropy measurements („binding competition“). Third, the method was employed for dynamic anisotropy enhancement allowing for the selective identification of cooperatively binding fragments. Lately, we have introduced a method for dynamic substrate enhancement for the targeting of secondary binding pockets of enzymes.
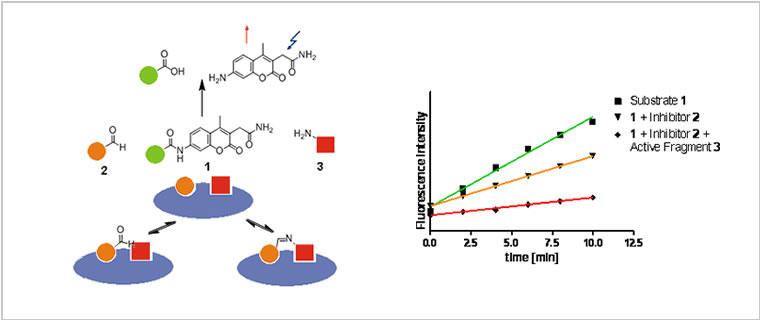 Fig. 1: The concept of Ligation Screening. Site-specific detection of fragments by substrate competition.
Fig. 1: The concept of Ligation Screening. Site-specific detection of fragments by substrate competition.
Chemical biology with fragment-based ligands
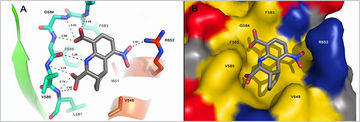 Bioactive molecules developed from fragment-based methods are characterized structurally and functionally in a number of projects.
Bioactive molecules developed from fragment-based methods are characterized structurally and functionally in a number of projects.
Inhibitors of the protein tyrosine phosphatase Shp2 which are active at nanomolar concentrations have been developed. These compounds have been demonstrated to be active in cellular models of metastasis cellular assays and block cellular mobility like cell scattering with no signs of toxicity. Therefore these molecules are currently validated in animal models in the framework of the pre-GoBio-competition. A second development project is funded by the Investitionsbank Berlin investigating inhibitors of the transcription faktor STAT5 towards leukaemic cells.
Linkages of active peptide fragments to especially potent structures is pursued in the framework of the SFB 765. There we work on multivalent, peptide-based constructs which target intracellular protein-protein interaction domains. Polymeric attachment of peptide drugs is employed in order to enhance their stability, cell permeability, and potency. Recently, multivalent enhancement was found by attachment of pro-apoptotic Bid-BH3 peptides. Peptide-polymer conjugates were also successfully used in imaging purposes.
Chemical synthesis of bioactive molecules
Projects of the group in all cases require a strong background in synthetic organic chemistry and are mostly based on our own developments of synthetic methodology. Traditionally, our synthetic focus is in solid phase and solid-supported synthesis of bioactive molecules, with strongholds in the preparation of peptide mimetics and carbohydrate derivatives. Over the years we have invented numerous reagents, some of these are commercially available to date.
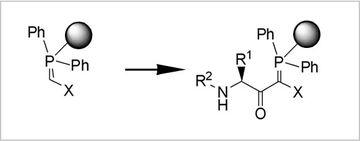 Fig.3: Polymer-supported C-Acylations
Fig.3: Polymer-supported C-Acylations
One specific goal in synthetic methodology is the integration of C-acylations in classical N-acylation (peptide) chemistry. Via C-acylations of polymeric phosphoranes, we have established access to a number of novel, reactive peptide derivatives which were useful for diverse ligation reactions and for the flexible variation of various biologically active protein ligands. For dynamic ligation assays as well as for the synthesis of Shp2-inhibitors C-acylation products proved to be valuable.
Starting the ChemBioNet
In order to strengthen Chemical Biology research in Germany, we have co-initiated the national collaboration network “ChemBioNet”. The ChemBioNet hosts an open screening platform at the FMP, Berlin. We have contributed to the composition of chemical libraries and offer fragment-ligation screens to the chemical biology community. In addition, the ChemBioNet has initiated the focus group “Chemical Biology” joining the Chemical Biology activities of Dechema, GDCh, GBM and DPhG.